Quantum Chemistry
Quantum chemistry maps molecular Hamiltonians onto qubits, enabling polynomial-time simulation of electron interactions
Source: mortalapps.com- Quantum chemistry simulations scale polynomially on quantum computers, compared to exponentially on classical systems.
- The molecular Hamiltonian must be mapped from fermionic operators to qubit Pauli operators using transformations like Jordan-Wigner.
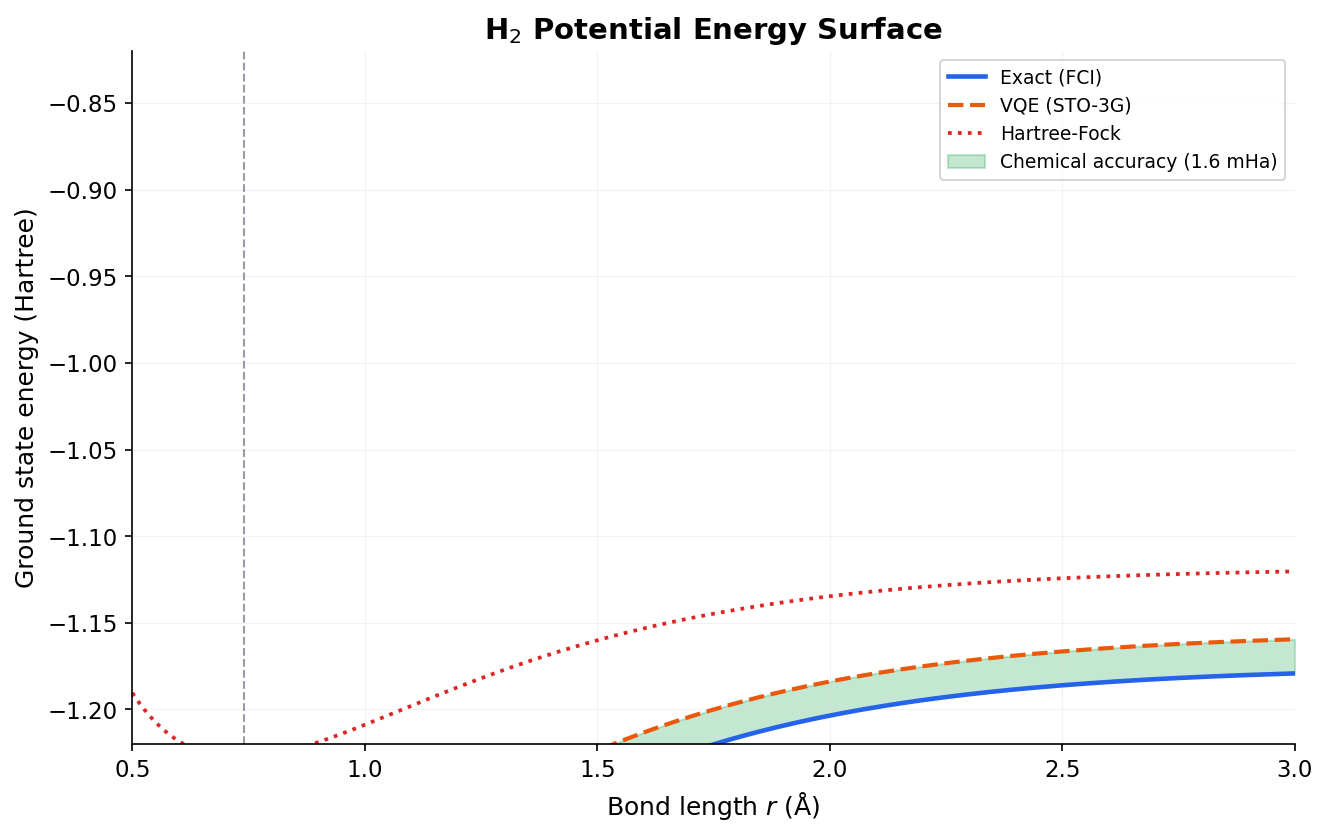
- The Variational Quantum Eigensolver (VQE) uses the variational principle to find molecular ground-state energies.
- Chemical accuracy is defined as finding the energy within 1 kcal/mol of the true value, which is required for practical chemical predictions.
- Simulating complex industrial catalysts like FeMoco could revolutionize global energy consumption by optimizing fertilizer production.
- Current hardware is limited to small toy molecules, with utility-scale chemistry requiring fault-tolerant systems.
Why This Matters
Quantum chemistry is widely considered the most promising domain for achieving early, high-impact quantum advantage. Simulating the behavior of molecules at the quantum level is incredibly difficult for classical computers because the complexity scales exponentially with the number of electrons. By mapping the quantum states of molecules directly onto the quantum states of qubits, we can bypass these classical limitations and simulate chemical reactions with unprecedented precision.
Core Intuition
To understand why classical computers struggle with chemistry, imagine trying to draw a highly detailed map of a turbulent ocean. A classical computer tries to calculate every single wave, current, and droplet individually, which quickly becomes overwhelming. A quantum computer, however, is made of the same 'fluid' as the ocean. Instead of calculating the waves, we let the quantum system naturally form the waves itself, and then we measure the result.
Another analogy is a lock-and-key mechanism. Finding the ground state of a molecule is like finding the exact shape of a key that fits a complex lock. Classical computers must guess millions of shapes one by one. A quantum computer can mold itself into the lock's shape using superposition and interference, finding the lowest-energy configuration (the perfect fit) much faster.
Visualization
Technical Explanation
The primary goal in quantum chemistry is to solve the electronic Schrödinger equation to find the ground-state energy of a molecular Hamiltonian $H$. In the second quantization framework, the electronic Hamiltonian is expressed in terms of creation ($a^\dagger_i$) and annihilation ($a_j$) operators:
$$H = \sum_{pq} h_{pq} a^\dagger_p a_q + \frac{1}{2} \sum_{pqrs} h_{pqrs} a^\dagger_p a^\dagger_q a_s a_r$$
where $h_{pq}$ and $h_{pqrs}$ are one- and two-electron integrals calculated classically. To map this Hamiltonian onto qubits, we use transformations like the Jordan-Wigner or Bravyi-Kitaev transformation, which map the fermionic operators to Pauli operators ($X, Y, Z, I$):
$$a^\dagger_j \rightarrow \frac{1}{2} (X_j - iY_j) \otimes Z_{j-1} \otimes \dots \otimes Z_0$$
Once mapped, we use the Variational Quantum Eigensolver (VQE) to find the ground-state energy. VQE relies on the variational principle, which states that the expectation value of a Hamiltonian for any trial state $| \psi(\theta) \rangle$ is always greater than or equal to the true ground-state energy $E_0$:
$$\langle \psi(\theta) | H | \psi(\theta) \rangle \ge E_0$$
By preparing a parameterized trial state (often using the Unitary Coupled Cluster ansatz, UCCSD) and measuring the expectation value, a classical optimizer can iteratively update $\theta$ to minimize the energy, converging toward $E_0$. While current NISQ devices can only simulate small molecules like $H_2$ or $LiH$, simulating larger molecules like FeMoco (the active site in nitrogenase, requiring ~50-100 logical qubits) could revolutionize fertilizer production and industrial chemistry.